Papers, communications and reviews… our recent published work is here.

Electrochemical CO2 reduction
Chemical Valorisation of Carbon Dioxide, Chap. 6 (Green Chemistry Series, RSC Publishing)
About this book The role of carbon dioxide in our changing climate is now hard to ignore, and many countries are making pledges to reduce or eliminate their carbon output. Chemical valorisation of carbon dioxide, as an alternative to sequestration, is likely to play an important part in reaching these targets, and as such is one of the fastest developing areas of green chemistry and chemical reaction engineering. Providing a comprehensive panorama of recent advances in the methods and technologies for chemical valorisation of carbon dioxide, this book is essential reading for anyone with an interest in sustainability and green chemistry. Both the technological improvements in traditional processes and new methods and concepts are discussed, including various (renewable) electricity-based methods, as well as novel catalytic, photocatalytic and biocatalytic approaches.
https://pubs.rsc.org/en/content/ebook/978-1-83916-407-1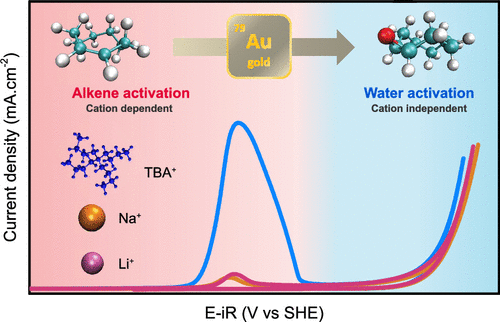
Controlling the Hydrophilicity of the Electrochemical Interface to Modulate the Oxygen-Atom Transfer in Electrocatalytic Epoxidation Reactions
J. Am. Chem. Soc. 144 (49), 22734–46, 2022
The electrocatalytic epoxidation of alkenes at heterogeneous catalysts using water as the sole oxygen source is a promising safe route toward the sustainable synthesis of epoxides, which are essential building blocks in organic chemistry. However, the physicochemical parameters governing the oxygen-atom transfer to the alkene and the impact of the electrolyte structure on the epoxidation reaction are yet to be understood. Here, we study the electrocatalytic epoxidation of cyclooctene at the surface of gold in hybrid organic/aqueous mixtures using acetonitrile (ACN) solvent. Gold was selected, as in ACN/water electrolytes gold oxide is formed by reactivity with water at potentials less anodic than the oxygen evolution reaction (OER). This unique property allows us to demonstrate that a sacrificial mechanism is responsible for cyclooctene epoxidation at metallic gold surfaces, proceeding through cyclooctene activation, while epoxidation at gold oxide shares similar reaction intermediates with the OER and proceeds via the activation of water. More importantly, we show that the hydrophilicity of the electrode/electrolyte interface can be tuned by changing the nature of the supporting salt cation, hence affecting the reaction selectivity. At low overpotential, hydrophilic interfaces formed using strong Lewis acid cations are found to favor gold passivation. Instead, hydrophobic interfaces created by the use of large organic cations favor the oxidation of cyclooctene and the formation of epoxide. Our study directly demonstrates how tuning the hydrophilicity of electrochemical interfaces can improve both the yield and selectivity of anodic reactions at the surface of heterogeneous catalysts.
https://doi.org/10.1021/jacs.2c10764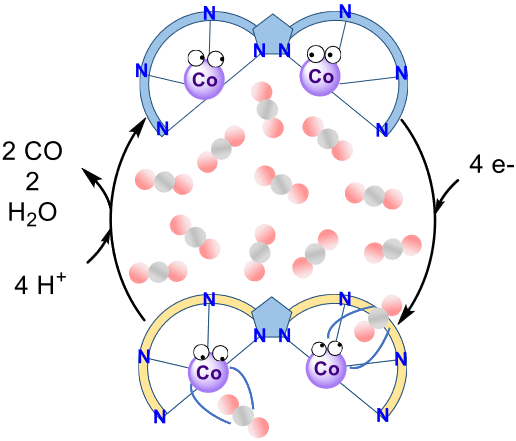
Electrocatalytic CO2 Reduction with a Binuclear bis-Terpyridine Pyrazole-Bridged Cobalt Complex
Chem. Eur. J., e20220236, 2023
A pyrazole–based ligand substituted with terpyridine groups at the 3 and 5 positions has been synthesized to form the dinuclear cobalt complex 1, that electrocatalytically reduces carbon dioxide (CO2) to carbon monoxide (CO) in the presence of Brønsted acids in DMF. Chemical, electrochemical and UV–vis spectro–electrochemical studies under inert atmosphere indicate pairwise reduction processes of complex 1. Infrared spectro–electrochemical studies under CO2 and CO atmosphere are consistent with a reduced CO–containing dicobalt complex which results from the electroreduction of CO2. In the presence of trifluoroethanol (TFE), electrocatalytic studies revealed single–site mechanism with up to 94% selectivity towards CO formation when 1.47 M TFE were present, at –1.35 V vs Saturated Calomel Electrode in DMF (0.39 V overpotential). The low faradaic efficiencies obtained (<50%) are attributed to the generation of CO–containing species formed during the electrocatalytic process, which inhibit the reduction of CO2.
https://doi.org/10.1002/chem.202202361
Graphene Wrapped Nickel Phthalocyanine Nanohybrid: Efficient Electrocatalyst for Nitrogen Reduction Reaction
Catal. Today, 423, 113938, 2023
Ammonia synthesis is a thermodynamically and kinetically demanding chemical process, and the senescent Haber–Bosch process has been the only way to manufacture ammonia under high pressure and temperature until recently. The electrochemical nitrogen reduction reaction (NRR) is a fungible method of ammonia production that requires strategic tailoring of a highly active, robust, and efficient electrocatalyst to overcome the challenges of high nitrogen (N2) bond dissociation energy, high specific surface area, more catalytic adsorption sites for N2, specificity to produce ammonia (NH3) as a major product, competitive hydrogen evolution reaction (HER), and long-term durability. Herein, Graphene wrapped nickel phthalocyanine (NiPc) nanohybrid (NiPc–RGO) was developed as an electrocatalyst, with a high ammonia yield of 23.9 µg h-1 mg-1cat and a Faradaic efficiency of 18.8% at –0.3 V vs. RHE, demonstrating outstanding durability and good selectivity. Isotopic labelling studies and numerous control trials are used to establish the validity of nitrogen sources in NH3 generation. These experimental results showed that NiPc-RGO hybrids could potentially serve as efficient NRR electrocatalysts for N2 fixation under ambient conditions.
https://doi.org/10.1016/j.cattod.2022.10.020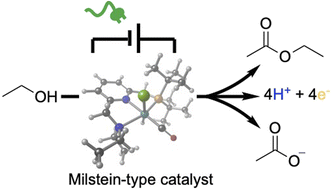
Electrification of a Milstein-type Catalyst for Alcohol Reformation
Chem. Sci. 13, 13220-13224, 2022
Novel energy and atom efficiency processes will be keys to develop the sustainable chemical industry of the future. Electrification could play an important role, by allowing to fine-tune energy input and using the ideal redox agent: the electron. Here we demonstrate that a commercially available Milstein ruthenium catalyst (1) can be used to promote the electrochemical oxidation of ethanol to ethyl acetate and acetate, thus demonstrating the four electron oxidation under preparative conditions. Cyclic voltammetry and DFT-calculations are used to devise a possible catalytic cycle based on a thermal chemical step generating the key hydride intermediate. Successful electrification of Milstein-type catalysts opens a pathway to use alcohols as a renewable feedstock for the generation of esters and other key building blocks in organic chemistry, thus contributing to increase energy efficiency in organic redox chemistry.
https://doi.org/10.1039/D2SC04533H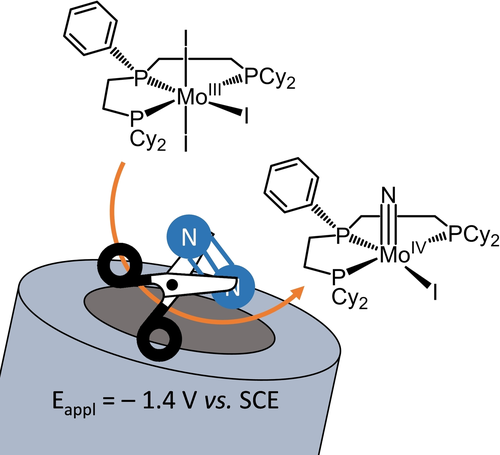
Molecular Electrochemical Reductive Splitting of Dinitrogen with a Molybdenum Complex
Angew. Chem. Int. Ed., e202209899, 2022
N2 splitting is achieved with a simple Mo complex at a carbon electrode. Controlled reduction (a 3 electron transfer process) at −1.4 V (vs. SCE) and ambient conditions (room T and atmospheric P) afforded a Mo nitride complex in 30 % yield. DFT studies support the transient formation of a MoI−N2-MoI bridging dimer.
https://doi.org/10.1002/anie.202209899
2022 Roadmap on Low Temperature Electrochemical CO2 Reduction
J. Phys. Energy, 4, 042003, 2022
Electrochemical CO2 reduction is an attractive option for storing renewable electricity and for the sustainable production of valuable chemicals and fuels. In this roadmap, we review recent progress in fundamental understanding, catalyst development, and in engineering and scale-up. We discuss the outstanding challenges towards commercialization of electrochemical CO2 reduction technology: energy efficiencies, selectivities, low current densities, and stability. We highlight the opportunities in establishing rigorous standards for benchmarking performance, advances in in operando characterization, the discovery of new materials towards high value products, the investigation of phenomena across multiple-length scales and the application of data science towards doing so. We hope that this collective perspective sparks new research activities that ultimately bring us a step closer towards establishing a low- or zero-emission carbon cycle.
https://doi.org/10.1088/2515-7655/ac7823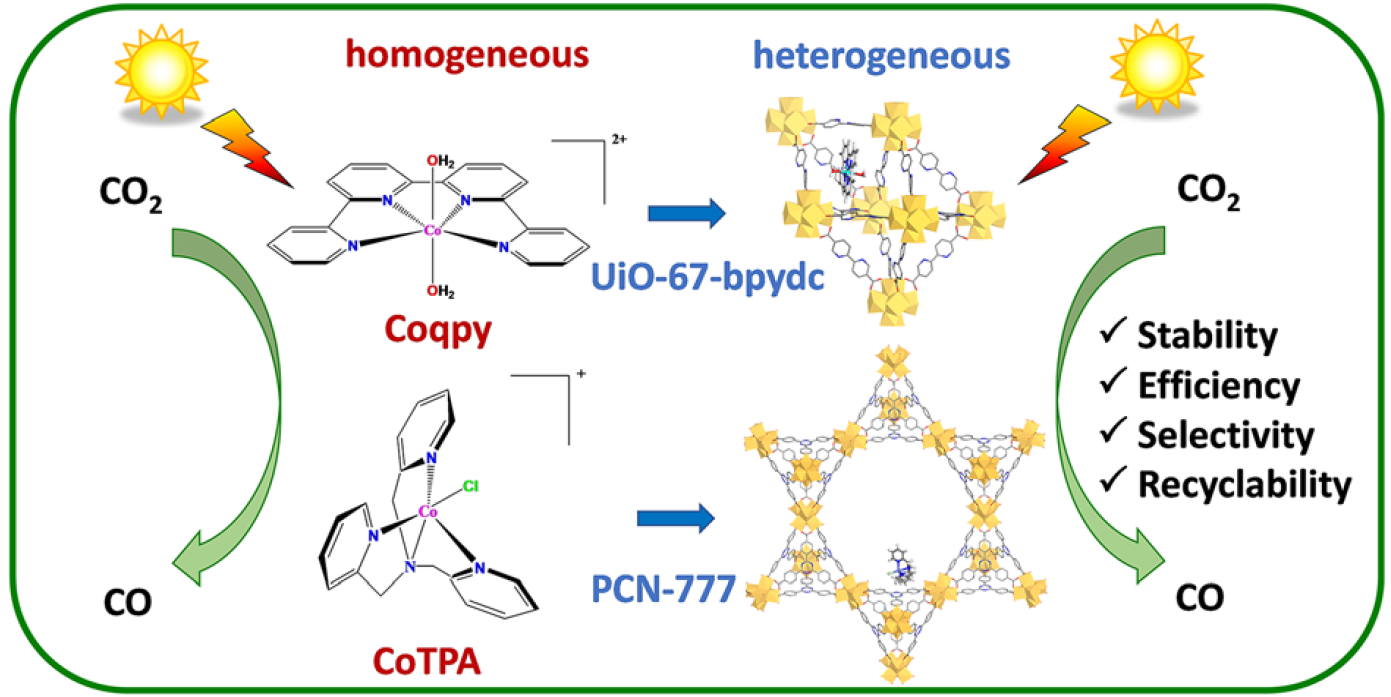
Heterogenization of Molecular Cobalt Catalysts in Robust Metal-Organic Frameworks for Efficient Photocatalytic CO2 Reduction
Catal. Sci. Technol., 12, 5418-5424, 2022
A Co-quaterpyridine (Coqpy) and a Co-tris(2-pyridylmethyl)amine cobalt (CoTPA) complexes, known for their ability to reduce CO2 to CO under visible-light irradiation in homogeneous conditions, were immobilized using the impregnation method in two different Zr-based metal-organic frameworks, UiO-67-bpydc and PCN-777, respectively. The successful synthesis of Coqpy@UiO-67-bpydc shows that linker deficiencies can allow catalytic complexes (Cat) to enter into the cavities of the Zr-based MOF while they would not in the defect-free MOF. The composites were characterized through solid state characterization techniques complemented by Density Functional Theory calculations in order to locate the catalysts into the MOF’s pores and estimate the host/guest interactions involved. The two noble-metal free Cat@MOF composites reduce CO2 to CO under visible-light irradiation in CH3CN/H2O (5:1) solutions, in the presence of [Ru(phen)3]2+ as solution-dissolved external photosensitizer and dimethylphenylbenzimidazoline (BIH) as electron and proton donor. CO yields, TON and selectivity reached 619 µmol g-1 h-1, 268 and 93% respectively for Coqpy@UiO-67-bpydc and 368 µmol g-1 h-1, 482 and 94% for CoTPA@PCN-777 after 24 h solar simulated illumination. These photocatalytic studies evidenced that the composite materials are efficient, selective and recyclable and that the MOF scaffold offers a favorable environment for constant CO production rate over a long time period.
https://doi.org/10.1039/d2cy01147f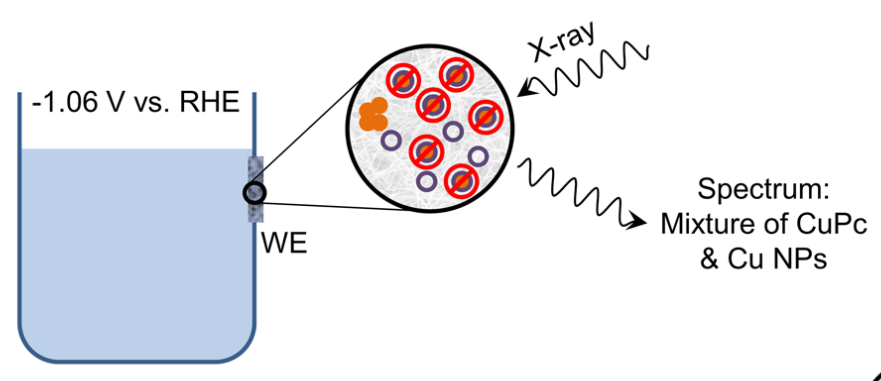
Confined Molecular Catalysts Provide an Alternative Interpretation to the Electrochemically Reversible Demetallation of Copper Complexes
Nat. Commun. 13, 4190, 2022
Metal phthalocyanines and porphyrins are among the most popular molecular catalysts for the electrochemical reduction of CO2. Recently, some copper-based complexes from these ligand families have been reported to promote the formation of methane and even ethylene at a high rate, an unprecedented property for a molecular catalyst1. More recently, operando X-ray Absorption Spectroscopy studies revealed that under cathodic conditions, small copper nanoparticles were forming from complex demetallation and were responsible for the catalysis2. The same studies claimed that the molecular Cu complexes are electrochemically reassembled when the electrode potential goes back to anodic value. Herein, we bring a different interpretation to the latter point, in accordance with all experimental data provided in the original article. Our interpretation accounts for the presence of electrochemically inert metal complexes confined inside the catalytic film and calls for a reassessment of some well-established views.
https://doi.org/10.1038/s41467-022-31661-1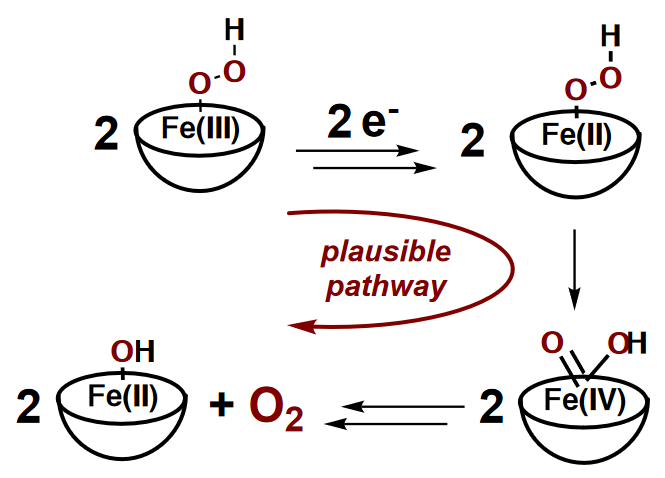
Heterolytic O-O Bond Cleavage Upon Single Electron Transfer to a Nonheme Fe(III)-OOH Complex
Chem. Eur. J. 28 (53), e202201600, 2022
The one-electron reduction of the nonheme iron(III)-hydroperoxo complex, [FeIII(OOH)(L52)]2+ (L52 = N -methyl- N , N’ , N’ -tris(2-pyridylmethyl)ethane-1,2-diamine), carried out at -70°C results in the release of dioxygen and in the formation of [FeII(OOH)(L52)]+ following a bimolecular process. This reaction can be performed either with cobaltocene as chemical reductant, or electrochemically. These experimental observations are consistent with the disproportionation of the hydroperoxo group in the putative FeII(OOH) intermediate generated upon reduction of the FeIII(OOH) starting complex. One plausible mechanistic scenario is that this disproportionation reaction follows an O-O heterolytic cleavage pathway via a FeIV-oxo species.
https://doi.org/10.1002/chem.202201600