Papers, communications and reviews… our recent published work is here.
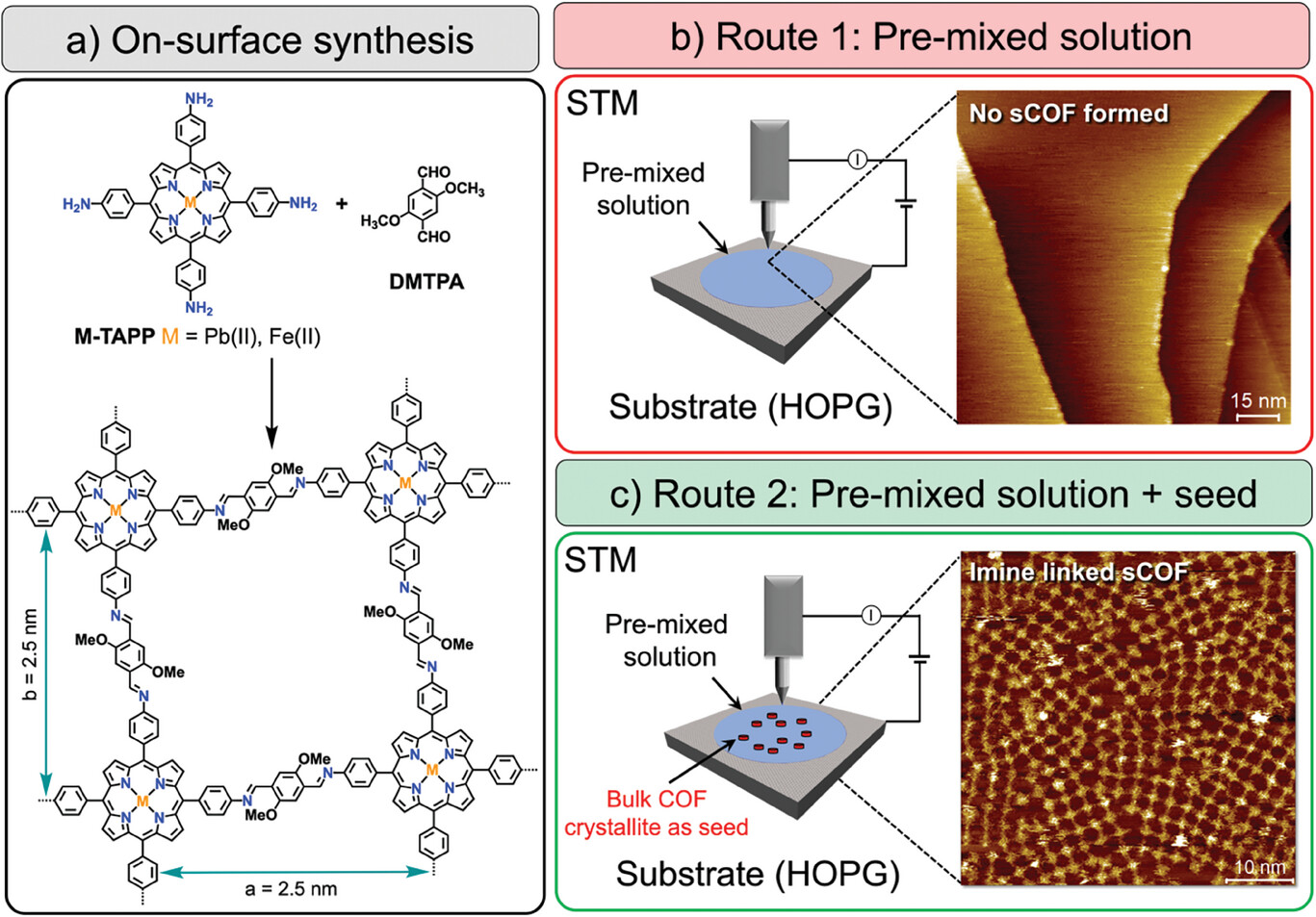
Single-Layered Imine-Linked Porphyrin-Based Two-Dimensional Covalent Organic Frameworks Targeting CO2 Reduction
Advanced Energy Materials, Early View, 2024
The reduction of carbon dioxide (CO2) using porphyrin-containing 2D covalent organic frameworks (2D-COFs) catalysts is widely explored nowadays. While these framework materials are normally fabricated as powders followed by their uncontrolled surface heterogenization or directly grown as thin films (thickness >200 nm), very little is known about the performance of substrate-supported single-layered (≈0.5 nm thickness) 2D-COFs films (s2D-COFs) due to its highly challenging synthesis and characterization protocols. In this work, a fast and straightforward fabrication method of porphyrin-containing s2D-COFs is demonstrated, which allows their extensive high-resolution visualization via scanning tunneling microscopy (STM) in liquid conditions with the support of STM simulations. The as-prepared single-layered film is then employed as a cathode for the electrochemical reduction of CO2. Fe porphyrin-containing s2D-COF@graphite used as a single-layered heterogeneous catalyst provided moderate-to-high carbon monoxide selectivity (82%) and partial CO current density (5.1 mA cm−2). This work establishes the value of using single-layered films as heterogene ous catalysts and demonstrates the possibility of achieving high performance in CO2 reduction even with extremely low catalyst loadings.
https://doi.org/10.1002/aenm.202304371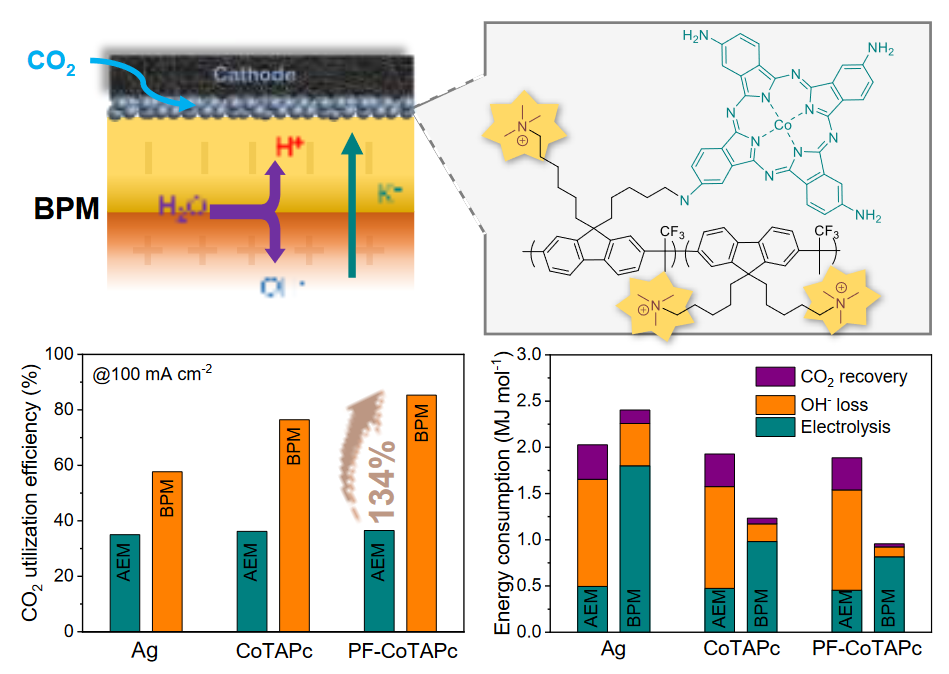
Backbone Engineering of Polymeric Catalysts for High-Performance CO2 Reduction in Bipolar Membrane Zero-Gap Electrolyzer
Angewandte Chemie Int. Ed., e202400414, 2024
Bipolar membranes (BPMs) have emerged as a promising solution for mitigating CO2 losses, salt precipitation and high maintenance costs associated with the commonly used anion-exchange membrane electrode assembly for CO2 reduction reaction (CO2RR). However, the industrial implementation of BPM-based zero-gap electrolyzer is hampered by the poor CO2RR performance, largely attributed to the local acidic environment. Here, we report a backbone engineering strategy to improve the CO2RR performance of molecular catalysts in BPM-based zero-gap electrolyzers by covalently grafting cobalt tetraaminophthalocyanine onto a positively charged polyfluorene backbone (PF-CoTAPc). PF-CoTAPc shows a high acid tolerance in BPM electrode assembly (BPMEA), achieving a high FE of 82.6% for CO at 100 mA/cm2 and a high CO2 utilization efficiency of 87.8%. Notably, the CO2RR selectivity, carbon utilization efficiency and long-term stability of PF-CoTAPc in BPMEA outperform reported BPM systems. We attribute the enhancement to the stable cationic shield in the double layer and suppression of proton migration, ultimately inhibiting the undesired hydrogen evolution and improving the CO2RR selectivity. Techno-economic analysis shows the least energy consumption (957 kJ/mol) for the PF-CoTAPc catalyst in BPMEA. Our findings provide a viable strategy for designing efficient CO2RR catalysts in acidic environments.
https://doi.org/10.1002/anie.202400414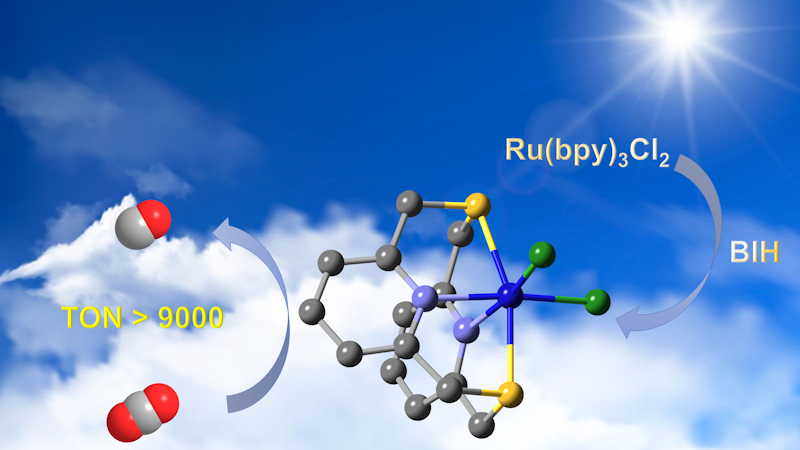
Efficient visible-light-driven carbon dioxide reduction by a bioinspired Nickel molecular catalyst
ChemSusChem, e202301892, 2024
Inspired by natural enzymes, this study presents a nickel-based molecular catalyst, [NiII(N2S2)]Cl2 (N2S2 = 2,11-dithia[3,3](2,6)pyridinophane), for the photochemical catalytic reduction of CO2 under visible light. The catalyst was synthesized and characterized using various techniques, including liquid chromatography-high resolution mass spectrometry (LC-HRMS), UV-Visible spectroscopy, and X-ray crystallography. The crystallographic analysis revealed a slightly distorted octahedral coordination geometry with a mononuclear Ni2+ cation, two nitrogen atoms and two sulfur atoms. Photocatalytic CO2 reduction experiments were performed in homogeneous conditions using the catalyst in combination with [Ru(bpy)3]Cl2 (bpy = 2,2’-bipyridine) as a photosensitizer and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as a sacrificial electron donor. The catalyst achieved a high selectivity of 89% towards CO and a remarkable turnover number (TON) of 7991 during 8 h of visible light irradiation under CO2 in the presence of phenol as a co-substrate. The turnover frequency (TOF) in the initial 6 h was 1079 h-1, with an apparent quantum yield (AQY) of 1.08%. Controlled experiments confirmed the dependency on the catalyst, light, and sacrificial electron donor for the CO2 reduction process. These findings demonstrate this bioinspired nickel molecular catalyst could be effective for fast and efficient photochemical catalytic reduction of CO2 to CO.
https://doi.org/10.1002/cssc.202301892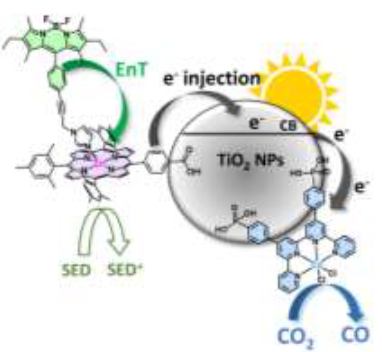
Antenna Effect in Noble Metal-Free Dye-Sensitized Photocatalytic Systems Enhances CO2-to-CO Conversion
Angewandte Chemie Int. Ed., e202318299, 2024
Dye-sensitized photocatalytic systems (DSPs) have been extensively investigated for solar-driven hydrogen (H2) evolution. However, their application in carbon dioxide (CO2) reduction remains limited. Furthermore, current solar-driven CO2-to-CO DSPs typically employ rhenium complexes as catalysts. In this study, we have developed DSPs that incorporate noble metal-free components, specifically a zinc-porphyrin as photosensitizer (PS) and a cobalt-quaterpyridine as catalyst (CAT). Taking a significant stride forward, we have achieved an antenna effect for the first time in CO2-to-CO DSPs by introducing a Bodipy as an additional chromophore to enhance light harvesting efficiency. The energy transfer from Bodipy to zinc porphyrin resulted in remarkable stability (turn over number (TON) = 759 vs. CAT), and high CO evolution activity (42 mmol g-1 h-1vs. CAT).
https://doi.org/10.1002/anie.202318299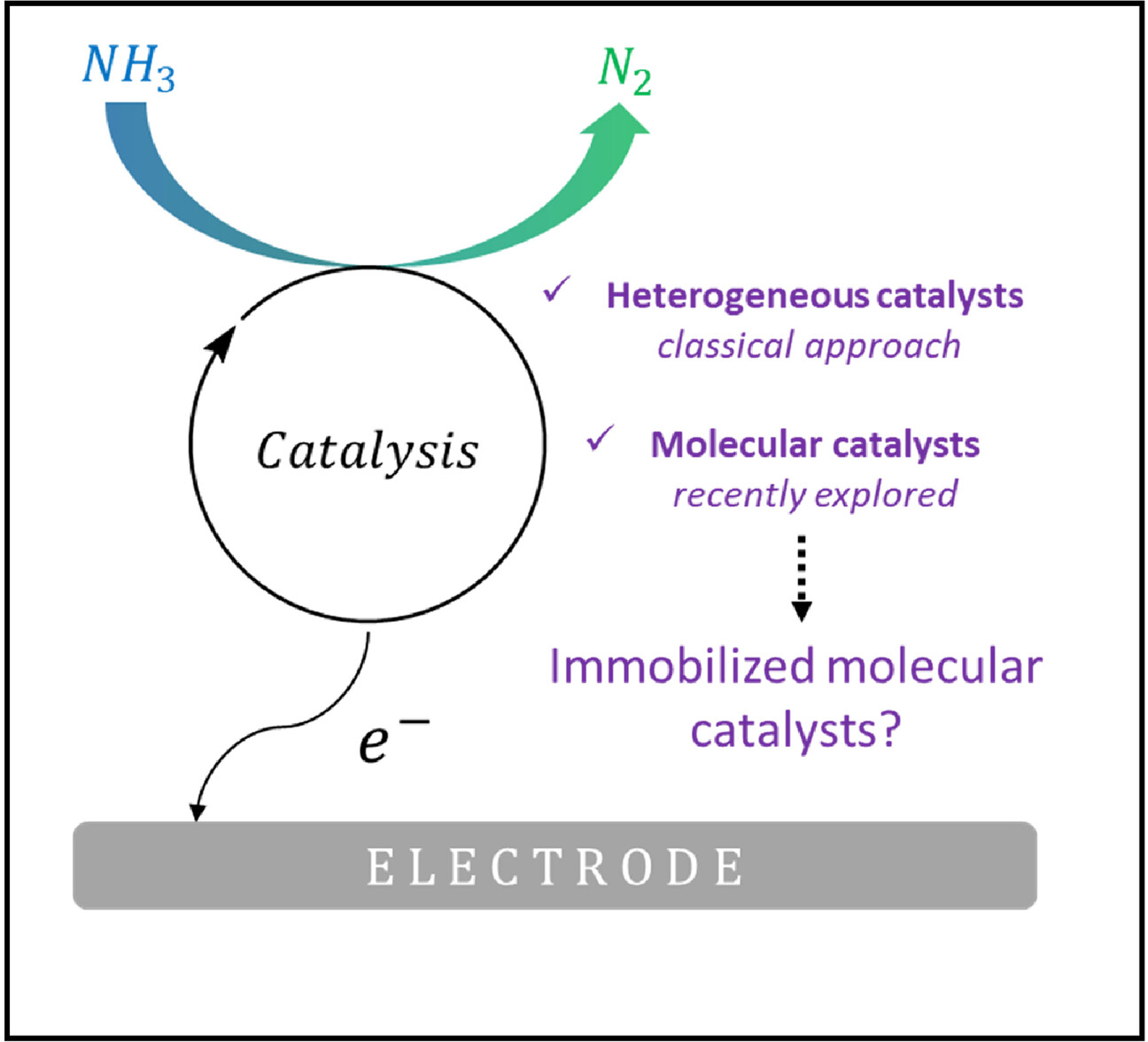
The Potential of Molecular Electrocatalysis for Ammonia-to-Dinitrogen Conversion
ChemElectroChem 11 (3), e202300462, 2024
Electrochemical ammonia oxidation reaction (eAOR) regains interest due to ammonia being an interesting alternative to hydrogen for fuel cell technologies. In the present review, we first discuss some of the most important findings on eAOR with solid catalysts, including mechanistic and feasibility aspects for practical implementation. We then examine the reports on molecular catalysis of eAOR that have recently emerged. We finally discuss immobilization strategies of these molecular catalysts, and discuss intrinsic advantages of those strategies, so as to guide the design of efficient catalytic systems able to compete with heterogeneous, solid catalysts.
https://doi.org/10.1002/celc.202300462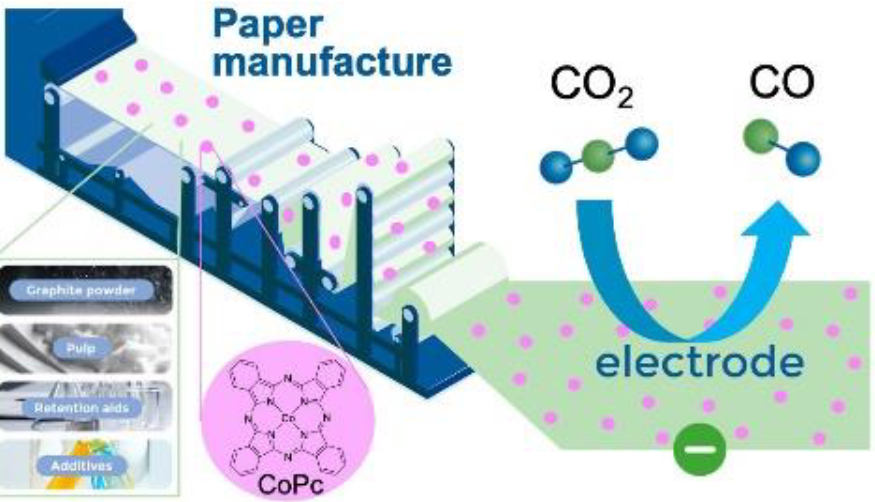
Electrocatalytic Functionalized Specialty Paper as Low Cost Porous Transport Layer Material in CO2-Electrolysis
ChemCatChem, e202300980, 2023
The development of low-cost and efficient electrolyzer components is crucial for practical electrochemical carbon dioxide reduction (ECR). In this study, facile non-woven cellulose-based porous transport layers (PTLs) were developed for high current density CO2-to-CO conversion. By depositing a cobalt phthalocyanine (CoPc) catalyst-layer over the PTLs, we fabricated ECR-functioning gas-diffusion-electrodes (GDEs) for both flow-cell and zero-gap electrolyzers. Under optimal conditions, the Faradaic Efficiency of CO (FECO) reached 92% at a high current density of 200 mA cm-2. Furthering the architecture of the GDEs, CoPc was incorporated into the initial PTL slurry, forming ECR-active PTLs without the need for an additional catalyst-layer. The new GDE-architecture favored the CoPc-distribution by enhancing the contact and interactions with the carbon substrate, and demonstrated a stable electrolysis process for over 50 h in a zero-gap cell at 200 mA cm-2 with an FECO of 80%.
https://doi.org/10.1002/cctc.202300980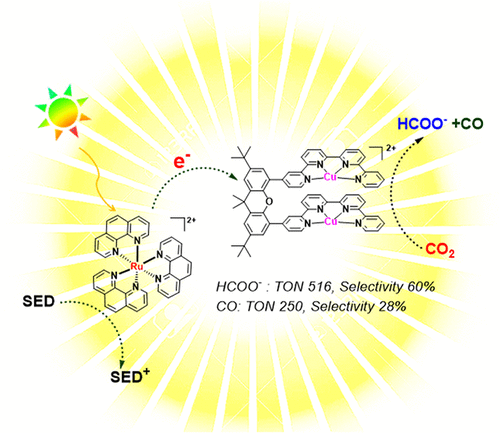
Visible-Light-Driven CO2 Reduction with Homobimetallic Complexes. Cooperativity between Metals and Activation of Different Pathways
J. Am. Chem. Soc. 145 (46), 25195–25202, 2023
Visible-light-driven reduction of CO2 to both CO and formate (HCOO–) was achieved in acetonitrile solutions using a homobimetallic Cu bisquaterpyridine complex. In the presence of a weak acid (water) as coreactant, the reaction rate was enhanced, and a total of ca. 766 TON (turnover number) was reached for the CO2 reduction, with 60% selectivity for formate and 28% selectivity for CO, using Ru(phen)22+ as a sensitizer and amines as sacrificial electron donors. Mechanistic studies revealed that with the help of cooperativity between two Cu centers, a bridging hydride is generated in the presence of a proton source (water) and further reacts with CO2 to give HCOO–. A second product, CO, was also produced in a parallel competitive pathway upon direct coordination of CO2 to the reduced complex. Mechanistic studies further allowed comparison of the observed reactivity to the monometallic Cu quaterpyridine complex, which only produced CO, and to the related homobimetallic Co bisquaterpyridine complex, that has been previously shown to generate formate following a mechanism not involving the formation of an intermediate hydride species.
https://doi.org/10.1021/jacs.3c07799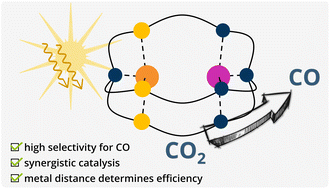
A CuICoII cryptate for the visible light-driven reduction of CO2
Chem. Sci., 14, 12774-12783, 2023
Among the rare bimetallic complexes known for the reduction of CO2, CoIICoII and ZnIICoII hexamine cryptates are described as efficient photocatalysts. In close relation to the active sites of natural, CO2-reducing enzymes, we recently reported the asymmetric cryptand {NSNN}m ({NSNN}m = N[(CH2)2SCH2(m-C6H4)CH2NH(CH2)2]3N) comprising distinct sulphur- and nitrogen-rich binding sites and the corresponding CuIMII (MII = CoII, NiII, CuII) complexes. To gain insight into the effect of metals in different oxidation states and sulphur-incorporation on the photocatalytic activity, we herein investigate the CuICoII complex of {NSNN}m as catalyst for the visible light-driven reduction of CO2. After 24 h irradiation with LED light of 450 nm, CuICoII-{NSNN}m shows a high efficiency for the photocatalytic CO2-to-CO conversion with 9.22 μmol corresponding to a turnover number of 2305 and a high selectivity of 98% over the competing H2 production despite working in an acetonitrile/water (4 : 1) mixture. Experiments with mononuclear counterparts and computational studies show that the high activity can be attributed to synergistic catalysis between Cu and Co. Furthermore, it was shown that an increase of the metal distance results in the loss of synergistic effects and rather single-sited Co catalysis is observed.
https://doi.org/10.1039/D3SC02679E
Merging Electrocatalytic Alcohol Oxidation with C-N Bond Formation by Electrifying Metal-Ligand Cooperative Catalysts
Chem. Sci. 14, 13437-13445, 2023
Electrification of thermal chemical processes could play an important role in creating a more energy efficient chemical sector. Here we demonstrate that a range of MLC catalysts can be successfully electrified and used for imine formation from alcohol precursors, thus demonstrating the first example of molecular electrocatalytic C-N bond formation.This novel concept allowed to increase energy efficiiency by an order of magnitude compared to thermal catalysis. Molecular EAO and the electrification of homogeneous catalysts can thus contribute to current efforts for the electrocatalytic generation of C-N bonds from simple building blocks.
https://doi.org/10.1039/D3SC03408A
1D/2D interface engineering of a CoPc–C3N4 heterostructure for boosting the nitrogen reduction reaction to NH3
Dalton Trans. 52, 15360-15364, 2023
Herein, we demonstrate the construction of a 1D/2D heterostructure of cobalt phthalocyanine (CoPc)–carbon nitride (C3N4) for electrochemical N2 reduction to NH3. Improved performance originates from the higher exposure of active surface sites. The electrochemical NRR performance showed an NH3 formation rate of 423.8 μg h−1 mgcat−1, a high faradaic efficiency (FE) of 33%, and stability for 20 h. This study provides a new strategy for designing a highly efficient 1D/2D electrocatalytic system for ammonia synthesis.
https://doi.org/10.1039/D3DT01790G