Papers, communications and reviews… our recent published work is here.
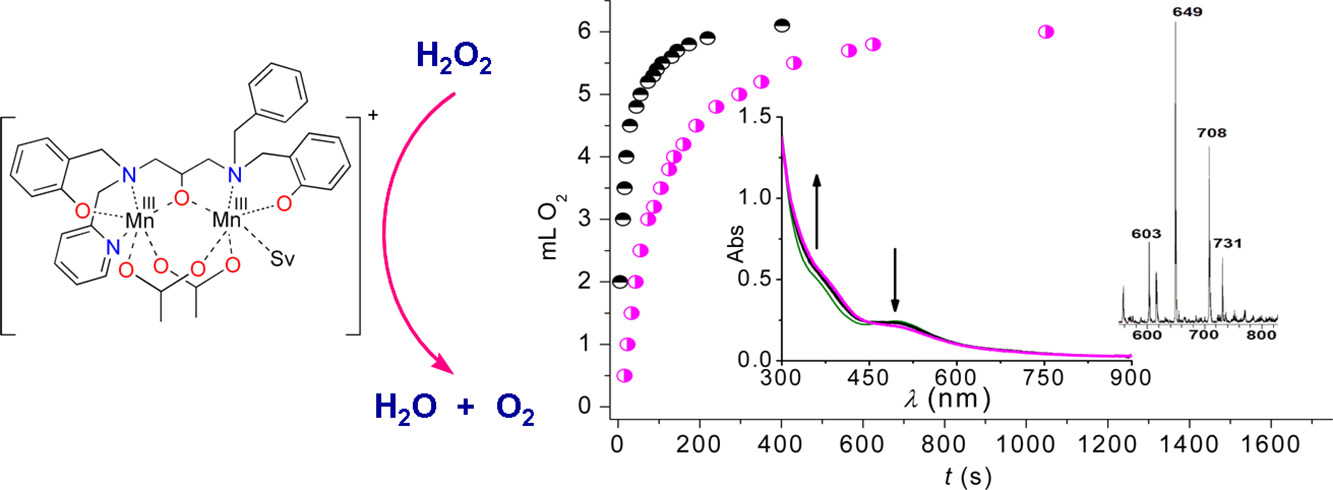
Functional Modeling of the MnCAT Active Site with a Dimanganese(III) Complex of an Unsymmetrical Polydentate N3O3 Ligand
J. Inorg. Biochem. 186, 10–16, 2018
A new diMnIII complex, [Mn2L(OAc)2(H2O)](BPh4)·3H2O (1), obtained with the unsymmetrical N3O3-ligand H3L = 1-[N-(2-pyridylmethyl),N-(2-hydroxybenzyl)amino]-3-[N′-(2-hydroxybenzyl),N′-(benzyl)amino]propan-2-ol, has been prepared and characterized. The unsymmetrical hexadentate ligand L3− leads to coordination dissymmetry (dissimilar donor atoms) around each Mn ion (N2O4 and NO4(solvent), respectively) leaving one labile site on one of the two Mn ions that facilitates interaction of the metal center with H2O2, as in Mn catalase. 1 is able to catalyze H2O2 disproportionation in acetonitrile, with second-order rate constant kcat = 23.9(2) M−1 s−1. The accessibility of the MnII2 state and the closeness of the two one-electron reduction processes suggest 1 employs MnIII2/MnII2 oxidation states for catalysis.
https://doi.org/10.1016/j.jinorgbio.2018.04.023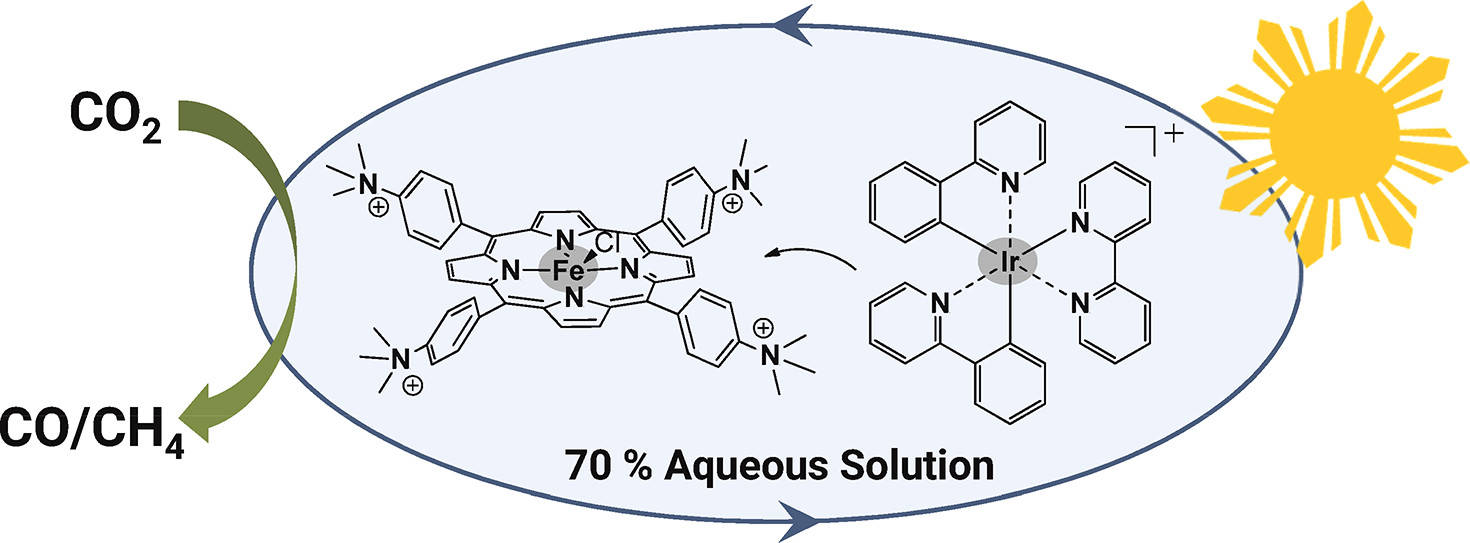
Toward Visible-Light Photochemical CO2-to-CH4 Conversion in Aqueous Solutions Using Sensitized Molecular Catalysis
J. Phys. Chem. C 122, 13834-13839, 2018
Solar fuels may be generated upon visible light induced catalytic reduction of carbon dioxide. This appealing approach remains highly challenging, especially when earth abundant catalysts, mild conditions, and water as a solvent were used. Employing an iron tetraphenyl porphyrin complex substituted by positively charged trimethylammonio groups at the para position of each phenyl ring and reduction with three electrons by the excited state of an iridium sensitizer (λ > 420 nm) reduce CO2 to CO and to CH4 in both acetonitrile and aqueous solutions (acetonitrile/water 3:7 v:v) with good selectivity. Stability of the catalytic system remains a weakness and the reasons were analyzed.
https://doi.org/10.1021/acs.jpcc.8b00950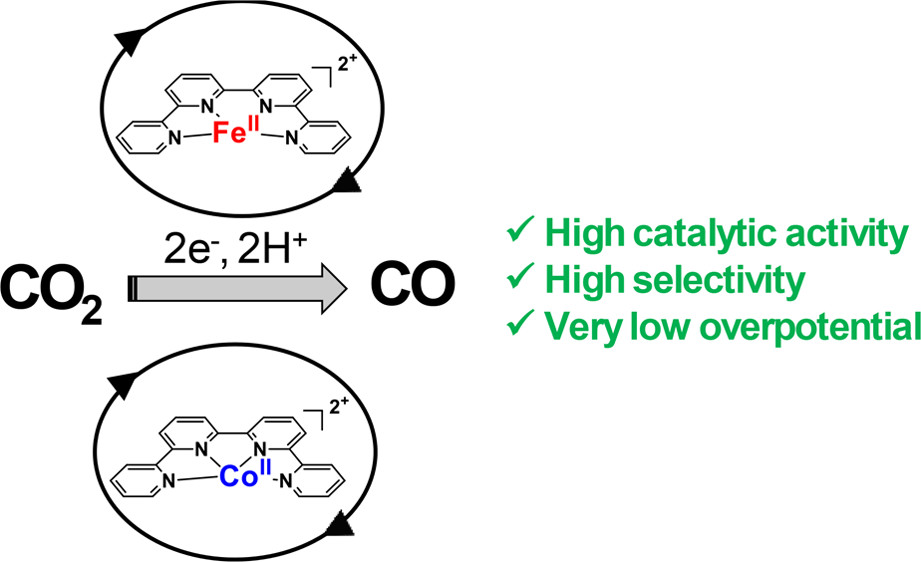
Highly Selective Molecular Catalysts for the CO2‑to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs Co Quaterpyridine Complexes upon Mechanistic Studies
ACS Catal. 8, 4, 3411-3417, 2018
[MII(qpy)(H2O)2]2+ (M = Fe, Co; qpy: 2,2′:6′,2″:6″,2‴-quaterpyridine) complexes efficiently catalyze the electrochemical CO2-to-CO conversion in acetonitrile solution in the presence of weak Brönsted acids. Upon performing cyclic voltammetry studies, controlled-potential electrolysis, and spectroelectrochemistry (UV–visible and infrared) experiments together with DFT calculations, catalytic mechanisms were deciphered. Catalysis is characterized by high selectivity for CO production (selectivity >95%) in the presence of phenol as proton source. Overpotentials as low as 240 and 140 mV for the Fe and Co complexes, respectively, led to large CO production for several hours. In the former case, the one-electron-reduced species binds to CO2, and CO evolution is observed after further reduction of the intermediate adduct. A deactivation pathway has been identified, which is due to the formation of a Fe0qpyCO species. With the Co catalyst, no such deactivation occurs, and the doubly reduced complex activates CO2. High scan rate cyclic voltammetry allows reaching kinetic conditions, leading to scan-rate-independent plateau-shaped voltammograms from which catalytic rate constant was obtained. The molecular catalyst is very active for CO production (turnover a frequency of 3.3 × 104 s–1 at 0.3 V overpotential), as confirmed by catalytic a Tafel plot showing a comparison with previous catalysts.
https://doi.org/10.1021/acscatal.7b04412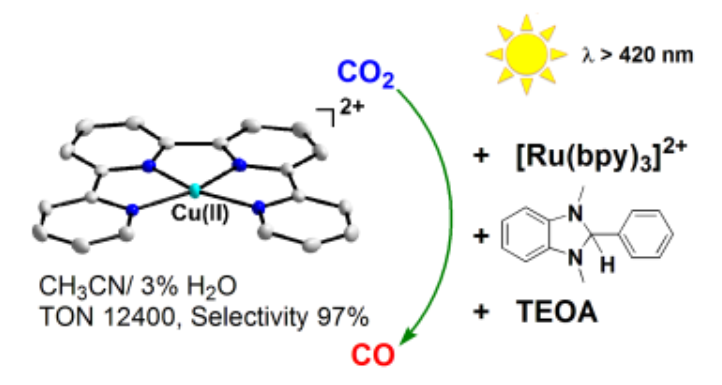
Photocatalytic Conversion of CO2 to CO by a Copper(II) Quaterpyridine Complex
ChemSusChem 10 (20), 4009-4013, 2017
The invention of efficient systems for the photocatalytic reduction of CO2 comprising earth-abundant metal catalysts is a promising approach for the production of solar fuels. One bottleneck is to design highly selective and robust molecular complexes able to transform the gas. The Cu(II) quaterpyridine complex [Cu(qpy)]2+ (1) is found to be a highly efficient and selective catalyst for visible-light driven CO2 reduction in CH3CN using [Ru(bpy)3]2+ as photosensitizer, BIH/TEOA as sacrificial reductant. The photocatalytic reaction is greatly enhanced by the presence of H2O (1–4% v/v), and a TON of >12,400 for CO production can be achieved with 97% selectivity, which is among the highest of molecular 3d CO2 reduction catalysts. Results from Hg-poisoning and dynamic light scattering (DLS) experiments suggest that this photocatalysis is homogenous. To the best of our knowledge 1 is the first example of molecular Cu-based catalyst for the photoreduction of CO2.
http://dx.doi.org/10.1002/cssc.201701354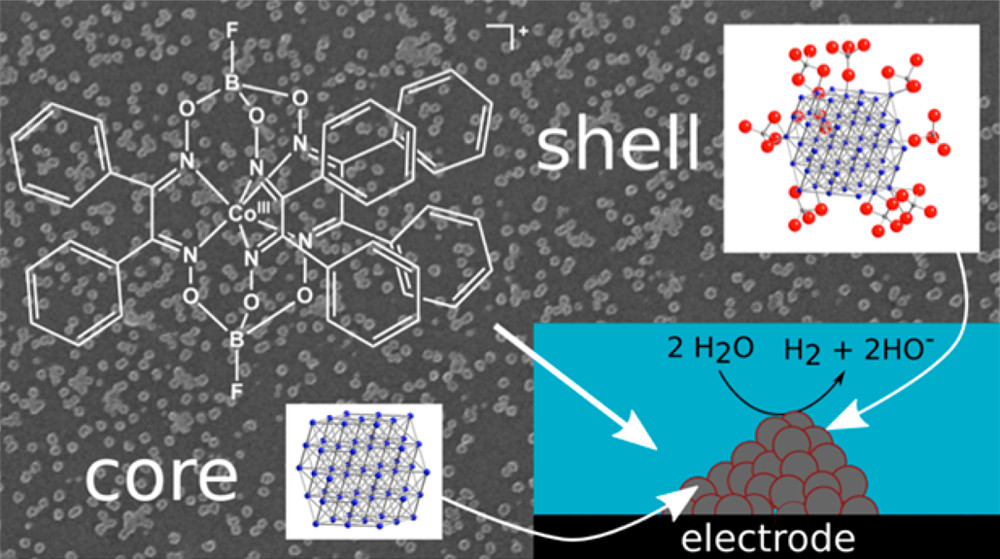
In Situ Observation of the Formation and Structure of Hydrogen-Evolving Amorphous Cobalt Electrocatalysts
ACS Energy Lett. 2 (11), 2545-2551, 2017
We have used in situ and operando X-ray absorption spectroscopy at the cobalt K-edge to study the formation of cobalt nanoparticles from a molecular precursor, as well as their structural evolution under hydrogen-evolving electrocatalytic conditions. We show that these particles, which are about 100−150 nm in diameter overall, are made of an uncommon form of amorphous metallic cobalt, the smallest ordered unit being 1 nm clusters of ∼50 cobalt atoms. In aqueous solution, these porous particles are partly oxidized into cobalt(II), a fraction of which remains present as an outer shell during hydrogen evolution electrocatalysis, even at very high cathodic potentials. Our operando measurements show that the activity of the particles is correlated to the oxidized layer thickness, a thinner layer exposing a larger fraction of the active metallic cobalt and leading to a higher activity. These findings expand our current understanding of the solid−liquid interface in hydrogen evolution catalytic species in neutral pH and suggest new directions for the improvement of hydrogen-evolving catalytic systems.
https://doi.org/10.1021/acsenergylett.7b00789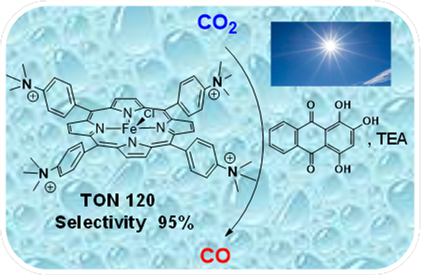
Visible-light Homogeneous Photocatalytic Conversion of CO2 to CO in Aqueous Solutions with an Fe Catalyst
ChemSusChem 10, 4447-4450, 2017
An iron-substituted tetraphenyl porphyrin bearing positively chargedtrimethylammonio groups at the para position of each phenyl ring catalyzes the photoinduced conversion of CO2.This complexiswater soluble and acts as amolecular catalyst to selectivelyreduce CO2 into CO under visible-light irradiation in aqueous solutions (acetonitrile/water=1:9 v/v) with the assistance of purpurin, a simple organic photosensitizer. CO is produced with acatalytic selectivity of 95% and turnover number up to 120, illustrating the possibility of photocatalyzing the reduction of CO2 in aqueous solutionbyusing visible light, a simple organic sensitizer coupled to an amine as a sacrificial electron donor,and an earth-abundant metal-based molecular catalyst.
https://doi.org/10.1002/ cssc.201701467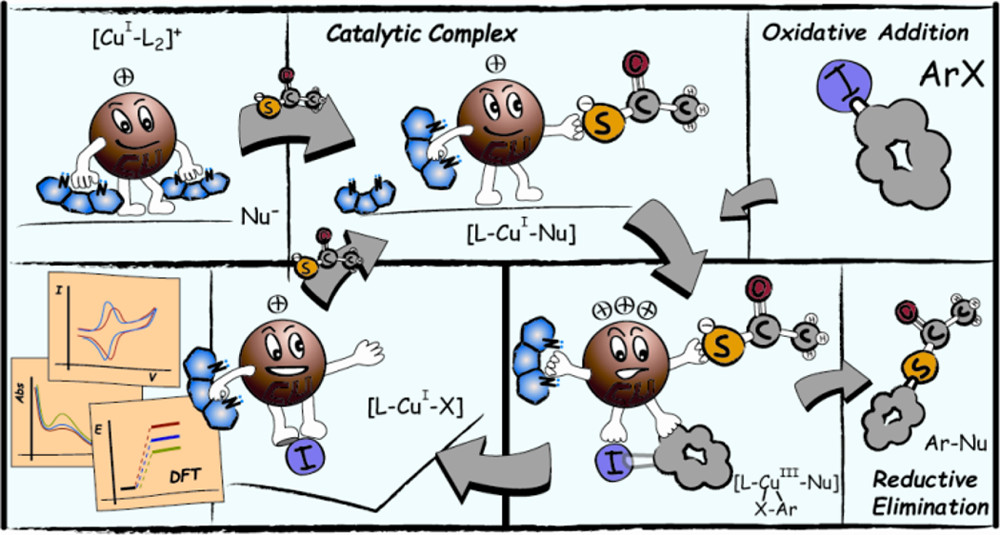
Mechanistic Insight into the Cu-Catalyzed C-S Cross-Coupling of Thioacetate with Aryl Halides. A Joint Experimental-Computational Study
J. Org. Chem. 82, 11464-11473, 2017
The mechanism of the Ullmann-type reaction between potassium thioacetate (KSAc) and iodobenzene (PhI) catalyzed by CuI associated with 1,10-phenanthroline (phen) as a ligand was explored experimentally and computationally. The study on C−S bond formation was investigated by UV−visible spectrophotometry, cyclic voltammetry, mass spec- trometry, and products assessment from radical probes. The results indicate that under experimental conditions the catalytically active species is [Cu(phen)(SAc)] regardless of the copper source. An examination of the aryl halide activation mechanism using radical probes was undertaken. No evidence of the presence of radical species was found during the reaction process, which is consistent with an oxidative addition cross-coupling pathway. The different reaction pathways leading to the experimentally observed reaction products were studied by DFT calculation. The oxidative addition−reductive elimination mechanism via an unstable CuIII intermediate is energetically more feasible than other possible mechanisms such as single electron transfer, halogen atom transfer, and σ-bond methatesis.
https://doi.org/10.1021/acs.joc.7b01991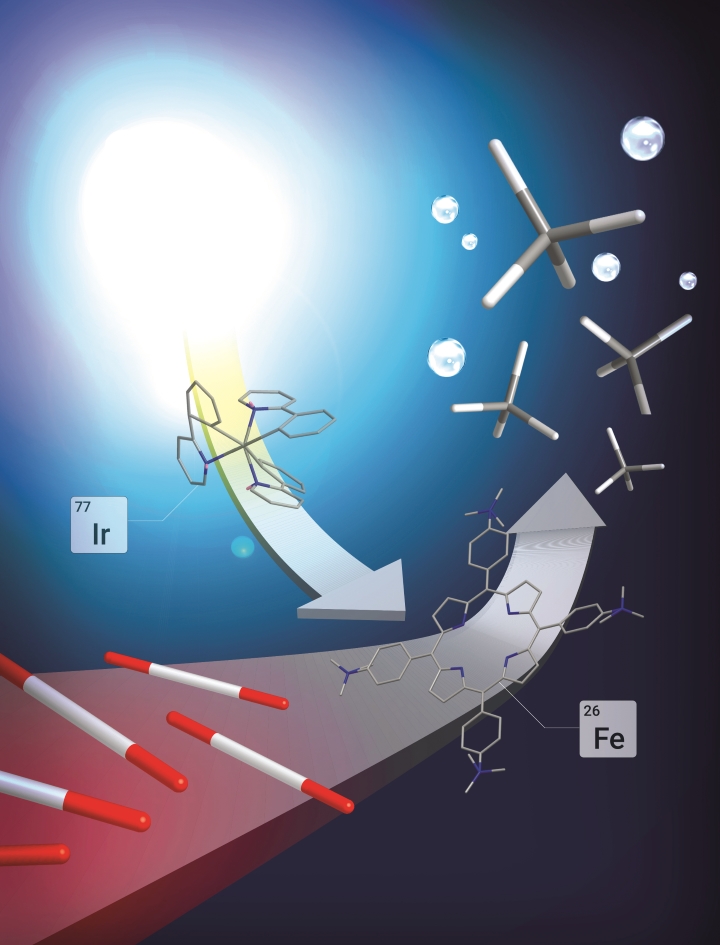
Visible-Light-Driven Methane Formation from CO2 with a Molecular Iron Catalyst
Nature 74 (548), 74-77, 2017
in principle reduce fossil fuel consumption and climate-changing CO2 emissions. One strategy aims for electrochemical conversions powered by electricity from renewable sources, but photochemical approaches driven by sunlight are also conceivable. A considerable challenge in both approaches is the development of efficient and selective catalysts, ideally based on cheap and Earth-abundant elements rather than expensive precious metals. Of the molecular photo- and electrocatalysts reported, only a few catalysts are stable and selective for CO2 reduction; moreover, these catalysts produce primarily CO or HCOOH, and catalysts capable of generating even low to moderate yields of highly reduced hydrocarbons remain rare. Here we show that an iron tetraphenylporphyrin complex functionalized with trimethylammonio groups, which is the most efficient and selective molecular electro- catalyst for converting CO2 to CO known, can also catalyse the eight-electron reduction of CO2 to methane upon visible light irradiation at ambient temperature and pressure. We find that the catalytic system, operated in an acetonitrile solution containing a photosensitizer and sacrificial electron donor, operates stably over several days. CO is the main product of the direct CO2 photoreduction reaction, but a twopot procedure that first reduces CO2 and then reduces CO generates methane with a selectivity of up to 82 per cent and a quantum yield (light-to-product efficiency) of 0.18 per cent. However, we anticipate that the operating principles of our system may aid the development of other molecular catalysts for the production of solar fuels from CO2 under mild conditions.
http://dx.doi.org/10.1038/nature23016
Molecular Catalysis of Electrochemical Reactions
Curr. Opin. Electrochem. 2 (1), 26-31, 2017
Molecular catalysis of electrochemical reactions currently attracts a lot of attention, notably concerning the transformation of small molecules in response to issues raised by modern energy challenges. This review summarizes recent advances in the mechanism analysis of multi-electrons–multi-steps processes involved in homogenous molecular catalysis of such electrochemical reactions. It also describes strategies for a rational catalyst benchmarking, through the establishment of “catalytic Tafel plots”. We show that careful analysis of through-structure and through-space substituent effects within a given family of catalyst is a powerful tool for intelligent design of new catalysts. Challenges raised by the “heterogenization” of molecular catalysts are discussed in the final section.
http:// dx.doi.org/ 10.1016/ j.coelec.2017.02.006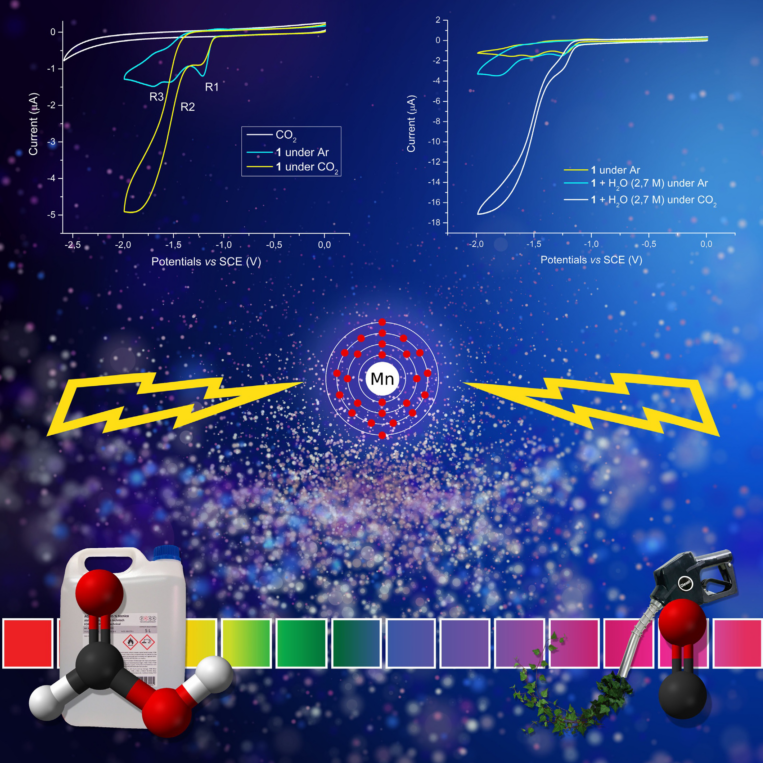
Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(bpy-R)(CO)3Br] Complexes
Chem. Eur. J. 23, 4782, 2017
The electrochemical behavior of fac‐[Mn(pdbpy)(CO)3Br] (pdbpy=4‐phenyl‐6‐(phenyl‐2,6‐diol)‐2,2′‐bipyridine) (1) in acetonitrile under Ar, and its catalytic performances for CO2 reduction with added water, 2,2,2‐trifluoroethanol (TFE), and phenol are discussed in detail. Preparative‐scale electrolysis experiments, carried out at −1.5 V versus the standard calomel electrode (SCE) in CO2‐saturated acetonitrile, reveal that the process selectivity is extremely sensitive to the acid strength, producing CO and formate in different faradaic yields. A detailed spectroelectrochemical (IR and UV/Vis) study under Ar and CO2 atmospheres shows that 1 undergoes fast solvolysis; however, dimer formation in acetonitrile is suppressed, resulting in an atypical reduction mechanism in comparison with other reported MnI catalysts. Spectroscopic evidence of Mn hydride formation supports the existence of different electrocatalytic CO2 reduction pathways. Furthermore, a comparative investigation performed on the new fac‐[Mn(ptbpy)(CO)3Br] (ptbpy=4‐phenyl‐6‐(phenyl‐3,4,5‐triol)‐2,2′‐bipyridine) catalyst (2), bearing a bipyridyl derivative with OH groups in different positions to those in 1, provides complementary information about the role that the local proton source plays during the electrochemical reduction of CO2.
https://doi.org/10.1002/chem.201605546